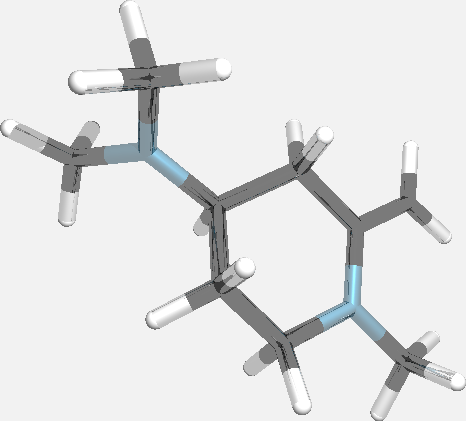
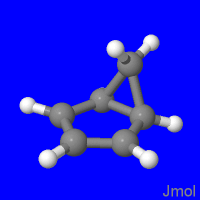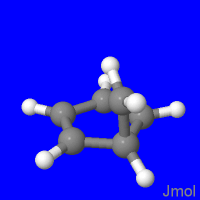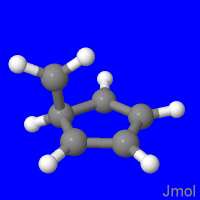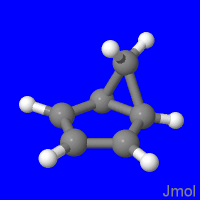
Ниже представлены анимации карусельной перегруппировки катиона C5H5CH2+, созданные с помощью программ jmol и gifsicle.


Скрипты написаны на Perl'е, все они читают как из файлов, данных в параметрах, так и из stdin'а, а также за несколькими исключениями (mldn, conformers, xyz -split) пишут на stdout преобразованный xyz, что позволяет организовывать конвееры с последовательными трансформациями.
script *.xyz cat *.xyz | script # То же самое, что и выше (почти всегда) script1 file.xyz | script2 > transformed_file.xyz
Все скрипты по script -h показывают справку.
Это простейший и наиболее распространенный формат пространственного строения молекул, его описание много где есть, например, тут и тут.
В одном xyz-файле можно хранить несколько структур (траектории оптимизации, сканирования, IRC, MD, набор конформеров, etc), причем достинается это простой конкатенацией отдельных xyz-файлов (надо только следить, чтобы не было лишних пустых строк).
Некоторые программы (например, orca, xtb, crest, MarvinSketch) и скрипты используют строки комментариев xyz-файлов, помещая туда энергию системы. Molden зачитывает эти энергии и показывает интерактивный график энергии по траектории. Это крайне удобно.
В своих скриптах зачитать энергии можно с помощью регулярного выражения, выкусывающего подстроку, похожую на число:
# Перл $comment_line =~ /(-?\d+\.\d+(?:[eE][+-]?\d+)?)/ && $energy = $1;
# Питон
import re
m = re.search('(-?\d+\.\d+(?:[eE][+-]?\d+)?)', comment_line)
energy = float(m.group(0))
Примеры comment_line:
-1.786388628385 Ammonia E0=-1.786388628385 energy: -178.6388628385e-02 gnorm: 0.000034641891
В xyz-файлах в блоках с координатами можно добавлять дополнительную 5-ю колонку чисел. Molden будет показывать эту цифирь около каждого атома (Меню Label - atom+charge). Вместо зарядов можно помещать туда другие параметры атома: хим. сдвиги ЯМР, неспаренную плотность, СТВ-константы ЭПР, etc. Удобно.
Это квантово-химический вьювер. На голых X-ах, поэтому имеет минимум зависимостей и работает на машинах с хотя бы базовой поддержкой графики (например, на удаленном кластере).
В molden'е необычная система координат: x вверх, y влево, z на нас.
Чтобы сделать как общепринято (x вправо, y вверх, z на нас)
xyz -a -ax file.xyz
Скрипт mldn - надстройка над molden'ом для работы в конвеере.
mldn file.xyz cat file.xyz | mldn # То же самое cat *.xyz | mldn # Все *.xyz как одна траектория
Некоторые программы и скрипты после оптимизации геометрии генерируют траектории оптимизации в виде конкатенированных xyz-файлов (например, orca file_trj.xyz, xtb xtbopt.log, pri2mol file.O.xyz). И часто нужно отредактировать эти файлы, оставив в них только последнюю, оптимизированную геометрию. Можно в редакторе, но если таких файлов десятки/сотни? На этот случай есть скрипт xyz.
xyz -i *.xyz
Опция -i означает редактирование файлов "по месту" (без неё идет печать на stdout).
Такой вызов скрипта подразумевает неявное использование опции -n=-1 (первая с конца точка траектории). С помощью этой опции можно специфицировать и другие точки (см. xyz -h).
Иногда требуется проредить траекторию. Для этого
xyz -thin=3 *.xyz # Печатать каждую третью точку
xyz -split trj.xyz # Разбивка траектории. Имена файлов - числа начиная с 0001 (формат %04d.xyz) xyz -split=name. trj.xyz # Имена файлов - по формату name.%04d.xyz
cat *.xyz > trj.xyz # Склеивание. Вместо cat можно различные скрипты
# Преобразование траектории с сортировкой по энергии (если последняя присутствует): conformers -only_gen trj.xyz cat ????.xyz > trj.xyz #rm ????.xyz
Нужно, в частности, для определения точек, в которых считать NICS (скрипт priin воспринимает XX именно в таком качестве).
addXX 1-6 file.xyz # Добавить фиктивный атом XX в центр атомов с 1 по 6-й
# (например, в центр бензольного кольца)
addXX '1-6(0,-1,1)' file.xyz # а также два дополнительных XX
# на расстоянии 1 ангстрем под и над кольцом.
Кроме того, добавление фиктивных атомов используется при переориентации молекулы (см. ниже).
Нужна для представления молекул в единообразном виде, чтобы их легко было сравнивать друг с другом сразу, без кручения во вьювере. Кроме того, часто используется для представления траекторий в пригодном для анимации виде.
align files.xyz | mldn # Подстройка каждой из структур к предыдущей (как в IRC)
# с последующим просмотром molden'ом
Если задана опция -n=N, то структуры подстраиваются не к предыдущей, а к N-ой. Ограничение align - все структуры должны иметь одинаковую нумерацию. Если это не так, то часть структур нужно перенумеровать (см. ниже).
xyz -a -ellips=2 files.xyz | mldn # Переориентация молекул по осям эллипсоида инерции
Переориентация по эллипсоиду удовлетворительно работает, если все структуры похожи по форме, но даже в этом случае бывает обращение направлений осей координат.
Ещё один способ переориентации - "по тройке атомов".
reorient -ijk=k,l,m files.xyz | mldn # Переориентация по тройке атомов с номерами k,l,m
Это, наверное, самый удачный и востребованный способ. Атомы из тройки не должны лежать на одной прямой, их логично выбирать из жесткой "центральной" части молекулы. В результате такой перенумерации зануляются следующие координаты:
x y z k: 0.00 0.00 0.00 Атом k в центре координат l: 0.00 0.00 Атом l на оси x m: 0.00 Атом m в плоскости xy
Некоторые примеры
# Соединить две разнонумерованные траектории без полной перенумерации одной из них: (reorient -ijk=k1,l1,m1 trj1.xyz; reorient -ijk=k2,l2,m2 trj2.xyz) | mldn
# К молекуле аммиака (1-й атом - азот) добавить XX в центр между протонами, # переориентировать с использованием XX и затем удалить XX addXX 2-4 | reorient -ijk=-1,2,3 | xyz -XX > NH3_reoriented.xyz
# Переориентировать молекулу, чтобы подчеркнуть её симметрию # (ориентация осей координат в стиле GAMESS). Используется внешняя программа symmetry xyz -symm=2 NH3.xyz | mldn
Ещё один пример, демонстрирующий пользу переориентации молекул в траектории и
зависимость способа переориентации от контекста. В этом примере используются фиктивные
атомы, задающие центр кольца и перпендикуляр к нему.
Ниже представлены анимации карусельной перегруппировки катиона
C5H5CH2+, созданные с помощью программ
jmol и
gifsicle.
Слева - анимация нативной IRC-траектории C5H5CH2+.xyz (RMSD между соседними структурами минимально). Траектории, относящиеся к следующим двум анимациям, получены последовательностью команд
addXX '1-5(0,1)' C5H5CH2+.xyz | reorient -ijk=-2,-1,1 | xyz -a -XX > C5H5CH2+_core_fix.xyz addXX 7,8 C5H5CH2+.xyz | reorient -ijk=6,-1,7 | xyz -a -XX > C5H5CH2+_CH2_fix.xyz
Вторая анимация показывает механизм перегруппировки в рамках концепции "мигрант-остов", предполагающей, что остов (в данном стучае - 5-членный цикл) неподвижен. В третьей анимацим неподвижна группа CH2. Не исключено, что и такое может понадобиться, например, в гипотетической ситуации, когда CH2 является частью полимера.
Перенумерация атомов в молекулах нужна, например, для объединения разнонумерованных траекторий, для сравнения наборов конформеров с разной нумерацией, etc. Можно переставлять строки в текстовом редакторе, но быстрее с помощью скрипта renum, которому нужно указать пермутационный вектор
renum 6,2-5 C6H6.xyz # поменять местами 1-й и 6-й атомы (6,2,3,4,5,1,7,...)
renum -p=1,6 C6H6.xyz # то же самое (попарная перестановка)
renum -auto trj.xyz # автоматическая перенумерация с помощью obabel. Образец для
# перенумерации берется из 1-й молекулы, т.е. ее нумерация не меняется
Перенумерация с помощью obabel происходит по топологии молекулы, поэтому подходит, например, для конформеров, но не годится для конкатенации IRC-траекторий реакций с разрывом/образованием связей. Более удобен скрипт, специально предназначенный для стыковки IRC-траекторий. Он каждую следующую траекторию перенумеровывает по последней точке предыдущей и при необходимости обращает ее конфигурацию. Нужно самому позаботиться, чтобы структуры на стыке траекторий совпадали. Например, имеем разнонумерованные траектории ircA.xyz и ircB.xyz двух последовательных реакций (последняя структура ircA.xyz совпадает с первой ircB.xyz).
join_IRC ircA.xyz ircB.xyz | mldn # Нумерация в ircB.xyz будет такая же, как в ircA.xyz
Скрипт мспользует другие скрипты: cfxyz, xyz, reflect, renum, align. Управляется опциями
-RMS если при стыковке RMS будет больше, то die. Default -RMS=0.2
-ref структура или начальная точка траектории, к которой подстраивать первую траекторию из аргументов.
Если не задано, то подстраивается к первой траектории из аргументов.
Бывает, что спуск по IRC с симметричного ПС в целях экономии времени посчитан только в одну сторону, В другую сторону можно достроить, но хочется сделать это так, чтобы получилась приятная глазу гладкая траектория, пригодная для анимации.
Возьмем траекторию irc0.xyz, начинающуюся с ПС и кончающуюся минимумом. В дополняющей траектории нужно поменять местами симметричные атомы (это проще с опцией -p в renum), затем сделать обращение конфигурации, реверсировать траекторию, выкинуть из неё точку с ПС и к тому что получится пристроить исходную траекторию.
renum -p='2,5 3,4 9,12 10,11' irc0.xyz | reflect | xyz -n=-1..2 > irc1.xyz align irc1.xyz irc0.xyz > irc.xyz
В данном примере минимумы тоже симметричны, поэтому можно дальше плясать уже от них. В итоге получится красивое движение по кругу, показанное выше.
Это был рассмотрен случай, когда ПС имеет симметрию Cs. Реже встречаются ПС с симметрией C2 и Ci. В случае Ci - всё то же самое. В случае же C2 отличие только в том, что не нужно делать обращение конфигурации. Например, имеем C2-симметричное ПС TS.xyz миграции протона в транс-дифторэтилене и одну из ветвей траектории IRC0.xyz (без ПС).
renum -p='1,2 4,7 5,6' IRC0.xyz > IRC1.xyz (xyz -r IRC0.xyz; cat TS.xyz IRC1.xyz) | align > IRC.xyz
Если ПС имеет более высокую симметрию, то нужно смотреть, симметрия какой из трех подгрупп (Cs, C2, Ci) нарушается при движеннии по координате реакции (мнимой моде гессиана) и действовать по алгоритмам для этих групп.
Замена знака у одной из координат x, y, z (отражение относительно плоскостей yz, xz, xy), а также замена знака у каждой из координат x, y, z (инверсия относительно центра) обращает конфигурацию молекулы. Замена знака у двух координат (вращение на 180° вокруг осей x, y, z) сохраняет конфигурацию молекулы.
reflect RSR.xyz > SRS.xyz # Обращение за счет отражения относительно плоскости xy
Пример из жизни. При генерировании конформеров хиральнной молекулы с помощью MarvinSketch наборы R.xyz и S.xyz, полученные из оптических изомеров, часто не совпадают, а иногда и вовсе почти не пересекаются. Объединить их в один набор, выкинув пересечения:
(cat R.xyz; reflect S.xyz) > t.xyz # Набор R-конформеров с дублями conformers -no_mopac temp.xyz # Удаление дублей cat temp.????.xyz > R_full.xyz # Конкатенация в полный набор rm t.xyz t.????.xyz # Удаление временных файлов
Иногда IRC обрывается, не доходя до минимума. В этом случае можно спуститься до минимума, запустив оптимизацию последней точки IRC и затем конкатенировать IRC-траекторию с траекторией оптимизации:
cat Opt.xyz >> IRC.xyz # Дописываем Opt в IRC
Но получается некрасиво, так как в траектории оптимизации в начале зачастую рывки, а в конце длинный хвост без видимых изменений. Чтобы получить более-менее гладкую пригодную для анимации траекторию:
xyz -a -irc=0.5 -avw=5 Opt.xyz >> IRC.xyz # Удаление из оптимизации точек, которые выше по энергии, чем предыдущая, а также точек, # в которых геометрия мало меняется (< 0.5 в единицах RMSD; логично брать шаг IRC). # Опция -avw задает сглаживание траектории
xyz -r trj.xyz | mldn # Просмотр траектории с точками, идущими в обратном порядке xyz -n=-1..1 trj.xyz | mldn # То же самое (xyz -r IRC0.xyz; cat TS.xyz IRC1.xyz) > IRC.xyz # Полная IRC-траектория (для Природы)
Очень востребованная задача. Для выявления одинаковых структур служит скрипт cfxyz, в котором реализованы три алгоритма.
1). Алгоритм Кабша (Kabsch),
представляющий собой метод вычисления оптимальной матрицы вращения,
которая минимизирует среднеквадратичное отклонение (RMSD) между двумя парными наборами точек.
Для этого алгоритма необходима одинаковая нумерация атомов в сравниваемых молекулах,
что достигается переориентацией молекул по главным осям их эллипсоидов инерции с последующей
перенумерацией одной из них по критерию пространственной близости пар атомов.
2). Модифицированный Grigoryan-Springborg algorithm,
сопоставляющий сортированные наборы межатомных расстояний однотипных атомов каждой из молекул.
3). Алгоритм, основанный на сопоставлении диэдральных углов,
заданных вручную либо автоматически с помощью программы dih_list.
Следующий пример иллюстрирует варианты использования cfxyz.
# Исспедуем ППЭ. Найденные ПС кладем в папку TS, минимумы - в min. # Нашли очередное ПС. Сначала проверяем, не было ли уже такого. cfxyz currTS.xyz vs. TS/*.xyz # Если currTS новое, то считаем IRC для него # Далее смотрим, с какими минимумами это ПС связано xyz -n=1,-1 IRC.xyz | cfxyz min/*.xyz # В таком варианте разделитель vs. не нужен
По завершению поиска можно построить диаграмму ППЭ с помощью скриптов xyz2levels/ levels2html или xyz2PES.
В наборе конформеров после DFT-оптимизации образуются дубли, которые нужно удалить.
cat *.xyz > confs.xyz # Объединяем оптимизированные структуры в траекторию conformers -no_ret -no_mopac confs.xyz # Сортировка по энергии и удаление дублей cat confs.????.xyz | mldn # Просмотр получившегося набора конформеров
Скрипт conformers сравнивает все конформеры из confs.xyz друг с другом. Единообразная нумерация не обязательна. С опцией -no_ret энантиомеры считаются одинаковыми. Опция -no_mopac предотвращает дополнительную оптимизацию полуэмпирикой или мол. механикой. Похожесть конформеров, которые считаются одинаковыми, регулируется опцией -RMS (по умолчанию -RMS=0.1). Если конформеров много, для ускорения сравнения можно использовать опции -ellips и -energy. Конформеры сразу будут считаться разными, если у них существенно различаются эллипсоиды инерции или энергии. Рекомендуется -ellips примерно как -RMS и -energy=0.001 (если энергия в хартри)
Иногда требуется провести небольшую однотипную модификацию серии молекул. Это можно делать с помощью скрипта subst.
Например, имеем молекулу бензола и генерируем производные:
subst -ii=7 -R=Me benzene.xyz > toluene.xyz subst -ii=H -R=F benzene.xyz > C6F6.xyz subst -ijk=1,7 -IJK=7,1 benzene.xyz benzene.xyz > biphenyl.xyz subst -ijk=8,2,4 -IJK=3,1,5 benzene.xyz benzene.xyz > naphthalene.xyz
Более подробно см. на страничке subst
Вращение вокруг заданной связи на заданный угол зачастую быстрее сделать скриптом в консоли, чем с помощью графического редактора структур, особенно это нужно сделать в серии молекул.
rotepi -rot=4-7 mol.xyz > mol1.xyz # Поворот вокруг связи 4-7
Умолчаемый угол поворота 180°. Регулируется опцией -angle=NNN.
Не работает, если заданная связь находится в составе цикла.
Бывает, что после поворота некоторые атомы накладываются.
Раздвинуть их можно опцией -xtb, которая вызывает программу xtb для небольшой
оптимизации структуры.
Более подробно см. на страничке rotepi
rotepi -epi=4 mol.xyz > mol2.xyz # Изменение конфигурации атома 4
Можно использовать опции -angle и -xtb (см. выше).
xyz -atoms=i,j,k,... *.xyz # Оставить в молекуле только перечисленные атомы xyz -atomsH=i,j,k,... *.xyz # вместе с присоединенными к ним атомами водорода xyz -noatoms=i,j,k,... *.xyz # Удалить из молекулы перечисленные атомы xyz -noatomsH=i,j,k,... *.xyz # вместе с присоединенными к ним атомами водорода xyz -H *.xyz # Удалить водороды xyz -XX *.xyz # Удалить фиктивные атомы
xyz -avw=n,p trj.xyz # "оконное" сглаживание - каждая точка представляется как среднее
# n окружающих. p - количество проходов, если отсутствует, то 1
Посмотреть, что получается, можно тут. Сглаживается геометрия и энергия.
cat reagent.xyz reactant.xyz | xyz -join > reagent+reactant.xyz
Энергетические параметры Energy HoF Edisp ZPE G суммируются. Молекулы ориентируются по осям эллипсоида инерции и сдвигаются вдоль наименьшей оси (z).
Такая операция имеет только вспомогательное значение, например, чтобы получить сумму реагентов в xyz-формате с суммарными энергиями, для скриптов barrier и xyz2levels. Или как заготовка для поиска вариантов "хорошего" взаиморасположения молекул программой crest
crest reagent+reactant.xyz
#!/bin/sh cp /usr/local/share/openbabel/2.4.1/babel_povray3.inc . perl -i -pe 's/#declare Bond_Thick_0 = \K0.1/0.15/; \ # Make bonds thicker s/#declare Color_F = rgbf<1.00,1.00,\K0.88/0.50/' \ # Different color for F babel_povray3.inc for file in $*; do echo $file xyz -i -ellips=2 -no_hide $file # Reorient xyz so that the atoms do not overlap name=`basename $file .xyz` obabel $file -O $name.pov -xm CST 2> /dev/null povray $name.pov 2> /dev/null mogrify -trim $name.png # Crop image pngquant -f -o $name.png $name.png # Compress image rm $name.pov done rm babel_povray3.inc
Нужен obabel и файл babel_povray3.inc из его дистрибутива, povray, скрипт xyz (необязательно, для автоматической переориентации молекул), mogrify (из ImageMagick, для обрезки лишнего фона), pngquant (необязательно, для уменьшения размера).
Для наглядной визуализации структуры нужно повернуть ее так, чтобы атомы не заслоняли друг друга. В автоматическом режиме это делает команда, которая обеспечивает такую ориентацию молекулы, при которой проекции атомов на плоскость xy меньше всего перекрываются.
xyz -i -ellips=2 -no_hide file.xyzОпция -ellips ориентирует молекулу по осям эллипсоида инерции (самая длинная ось x, самая короткая z).
Получается примерно так:
было 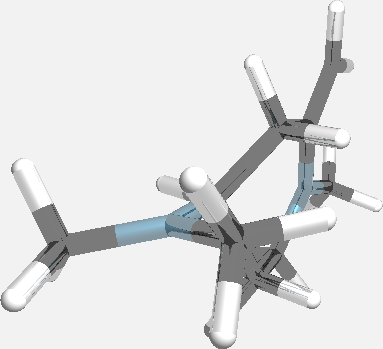 стало
стало 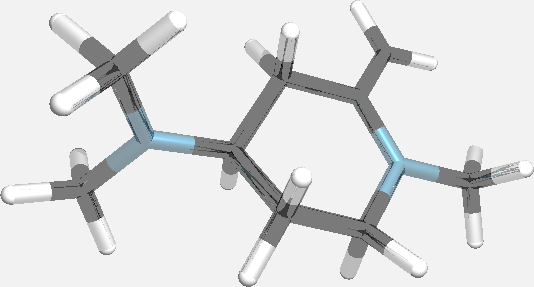 без -no_hide
без -no_hide 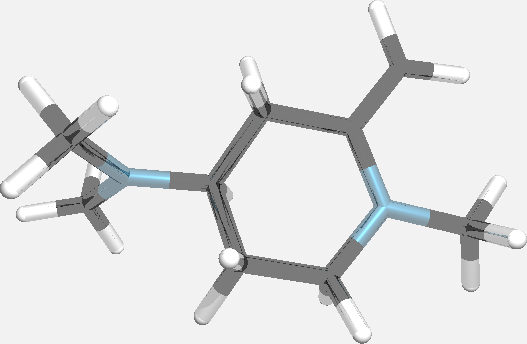 без -ellips
без -ellips